Research
Research Statement Summary
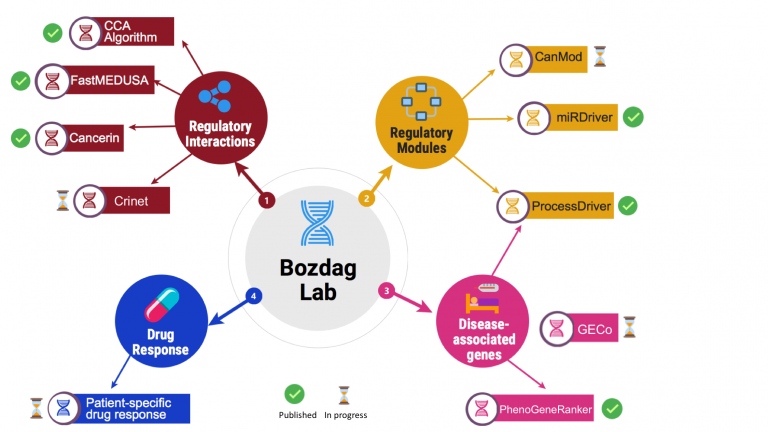
My lab's research goal is to develop open source integrative computational tools to analyze high dimensional biological, clinical and environmental exposure datasets to infer context-specific gene regulatory interactions and modules, and to predict disease associated genes and patient-specific drug response. With the advances in high throughput technologies in biology, numerous national and international consortiums have generated a vast amount of genotype, phenotype, gene expression, and epigenetic data (collectively called multi-omics data), which have been made available to the scientific community. Furthermore, ongoing large initiatives such as UK Biobank, Million Records Project, and All of Us research program will bring vast amounts of multi-omics datasets from millions of individuals. Each of these different data modalities (e.g., mRNA expression, DNA methylation, mutation, microRNA (miRNA) expression, and copy number alteration) describes one facet of the underlying biology. Consequently, there is a tremendous need for scalable methods that can integrate different layers of multi-omics datasets across millions of individuals from different backgrounds. These methods would produce valuable insights into human diseases and pave the way for precision medicine. My research program is devoted to developing integrative computational tools utilizing artificial intelligence, machine learning and data mining methods to analyze these multi-omics datasets.
FUNDING
Research Themes
1. Building machine learning models integrating multi-modal biomedical datasets
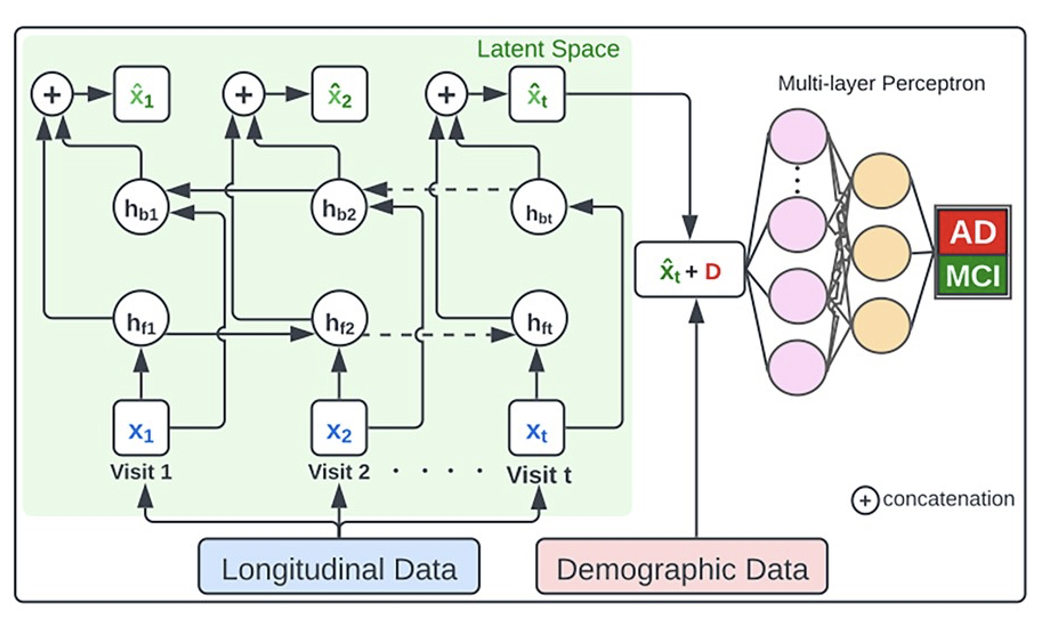
We have been involved in building several classification tools to predict gene expression, disease subtype, and drug carcinogenicity. Specifically, we have developed deep learning architecture called PPAD to integrate cross-sectional and longitudinal datasets of Alzheimer's disease to predict its progression. We developed a k-nearest neighbor-based model to predict gene expression changes based on DNA methylation signal. We employed ensemble-based learning method to predict in vivo carcinogenicity. We also developed tools to predict subtype of cancer patients, namely predicting epigenetic subtype and molecular subtype of glioblastoma patients. We also developed a tool named NRPReTo to predict nuclear receptor proteins based on sequence- and structure-based features.
Selected Publications
M. Talo and S. Bozdag, “ProtFun: A Protein Function Prediction Model Using Graph Attention Networks with a Protein Large Language Model,” May 17, 2025, GLBIO 2025, (Preprint: https://doi.org/10.1101/2025.05.13.653854)
C. Ozdemir, M. A. Olaimat, and S. Bozdag, “A Dynamic Model for Early Prediction of Alzheimer’s Disease by Leveraging Graph Convolutional Networks and Tensor Algebra,” Pac Symp Biocomput (PSB 2025), vol. 30, pp. 675–689, 2025. https://doi.org/10.1142/9789819807024_0048
M. A. Olaimat and S. Bozdag, “CAAT-EHR: Cross-Attentional Autoregressive Transformer for Multimodal Electronic Health Record Embeddings,” Jan. 31, 2025, arXiv: arXiv:2501.18891. https://doi.org/10.48550/arXiv.2501.18891.
Susman A, Krishnamurthy R, Li YC, Olaimat M, Bozdag S, Varghese B, Sheikh-Bahei N, Pandey G. Longitudinal Ensemble Integration for sequential classification with multimodal data. arXiv; 2024. http://arxiv.org/abs/2411.05983
M. T. Rahman, F. Saeed, S. Bozdag, and Alzheimer’s Disease Neuroimaging Initiative, “Identifying Alzheimer’s disease-associated genes using PhenoGeneRanker,” bioRxiv, p. 2024.11.12.623269, Nov. 2024, https://doi.org/10.1101/2024.11.12.623269.
S. S. Madugula, S. Pandey, S. Amalapurapu, and S. Bozdag, “NRPreTo: A Machine Learning-Based Nuclear Receptor and Subfamily Prediction Tool,” ACS Omega, May 2023, https://doi.org/10.1021/acsomega.3c00286.
M. Al Olaimat, J. Martinez, F. Saeed, S. Bozdag, and Alzheimer's Disease Neuroimaging Initiative, “PPAD: a deep learning architecture to predict progression of Alzheimer's disease,” (ISMB/ECCB 2023) Bioinformatics, vol. 39, no. Supplement_1, pp. i149-i157, Jun. 2023, doi: 10.1093/bioinformatics/btad249.
T. Yang, M. A. Al-Duailij, S. Bozdag and F. Saeed, Classification of Autism Spectrum Disorder Using rs-fMRI data and Graph Convolutional Networks, 2022 IEEE International Conference on Big Data (Big Data), Osaka, Japan, 2022, pp. 3131-3138, https://doi.org/10.1109/BigData55660.2022.10021070
Baur, B., & Bozdag, S. (2016). A Feature Selection Algorithm to Compute Gene Centric Methylation from Probe Level Methylation Data. PloS One, 11(2), e0148977. https://doi.org/10.1371/journal.pone.0148977
Baysan, M., Bozdag, S., Cam, M. C., Kotliarova, S., Ahn, S., Walling, J., … Fine, H. A. (2012). G-cimp status prediction of glioblastoma samples using mRNA expression data. PLoS One, 7(11), e47839. https://doi.org/10.1371/journal.pone.0047839
2. Graph representation learning methods for network biology
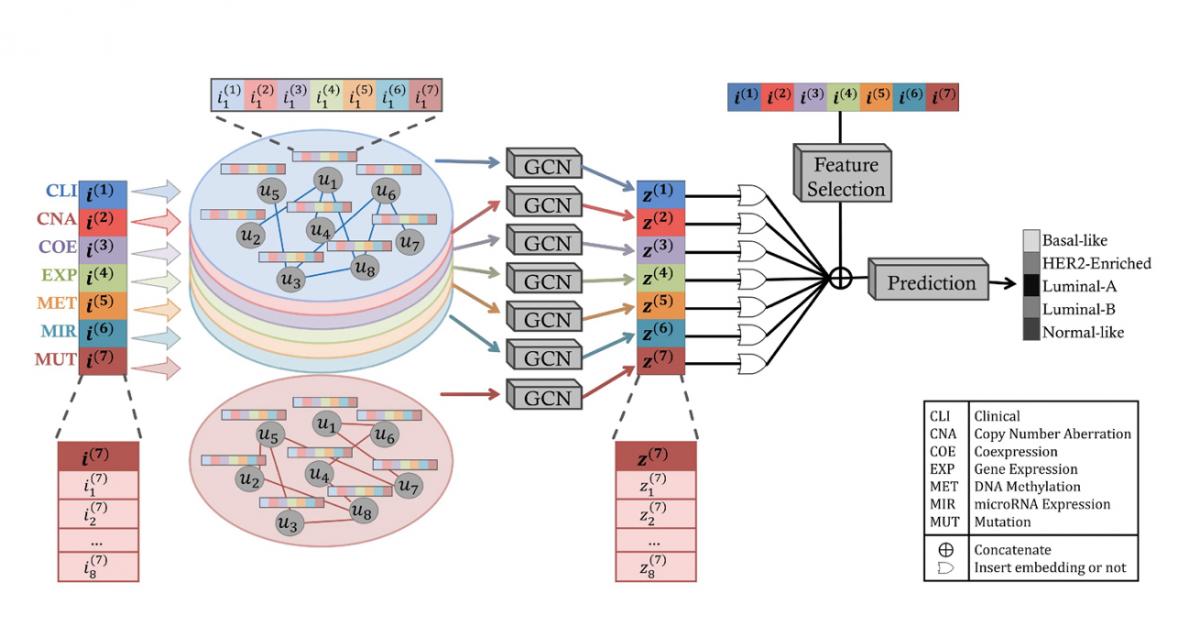
In recent years, we have developed several machine learning tools in network biology to integrate multi-modal biomedical datasets. We developed a random walk with restart method called PhenoGeneRanker that works on multiplex heterogeneous networks of genes and phenotypes (i.e., networks with multiple types of nodes and edges) to rank disease-associated genes. We have developed a node embedding method for multiplex heterogeneous networks called NeCO and used it to compute hypertension-related genes. We developed SUPREME, which integrates multi-layer networks for downstream tasks such as node classification and GRAF, which fuses multi-layer networks into a single network and perform downstream tasks.
Selected Publications
M. Talo and S. Bozdag, “Top-DTI: Integrating Topological Deep Learning and Large Language Models for Drug Target Interaction Prediction,” ISMB/ECCB 2025 (Bioinformatics) 2025 Jul 1;41(Supplement_1):i133–i141. https://doi.org/10.1093/bioinformatics/btaf183.
C. Ozdemir, Y. Vashishath, S. Bozdag, and Alzheimer’s Disease Neuroimaging Initiative, “IGCN: integrative graph convolution networks for patient level insights and biomarker discovery in multi-omics integration,” Bioinformatics, vol. 41, no. 6, p. btaf313, 2025, https://doi.org/10.1093/bioinformatics/btaf313.
B. Bose, B. Stranger, and S. Bozdag, “A network model for patient-derived drug response in breast cancer integrating multi-omics datasets,” Jun. 12, 2025, bioRxiv. https://doi.org/10.1101/2025.06.09.658757.
Zitnik M, Li MM, Wells A, Glass K, Gysi DM, Krishnan A, Murali TM, Radivojac P, Roy S, Baudot A, Bozdag S, Chen DZ, Cowen L, Devkota K, Gitter A, Gosline S, Gu P, Guzzi PH, Huang H, Jiang M, Kesimoglu ZN, Koyuturk M, Ma J, Pico AR, Pržulj N, Przytycka TM, Raphael BJ, Ritz A, Sharan R, Shen Y, Singh M, Slonim DK, Tong H, Yang XH, Yoon BJ, Yu H, Milenković T. Current and future directions in network biology. arXiv; 2023. http://arxiv.org/abs/2309.08478.
Z. N. Kesimoglu and S. Bozdag, “SUPREME: multiomics data integration using graph convolutional networks,” NAR Genomics and Bioinformatics, vol. 5, no. 2, p. lqad063, Mar. 2023, doi: 10.1093/nargab/lqad063.
Z. N. Kesimoglu & S. Bozdag (2023). GRAF: Graph Attention-aware Fusion Networks. arXiv preprint arXiv:2303.16781. https://doi.org/10.48550/arXiv.2303.16781
Dursun C, Kwitek A, Bozdag S. PhenoGeneRanker: Gene and Phenotype Prioritization Using Multiplex Heterogeneous Networks. IEEE/ACM Trans Comput Biol Bioinform. 2021 Jul 20;PP. PMID: 34283720 https://doi.org/10.1109/TCBB.2021.3098278
Dursun C, Smith JR, Hayman GT, Kwitek AE, Bozdag S. NECo: A node embedding algorithm for multiplex heterogeneous networks. 2020 IEEE International Conference on Bioinformatics and Biomedicine (BIBM). 2020. p. 146-149. https://doi.org/10.1109/BIBM49941.2020.9313595
3. Analysis of high dimensional biological datasets to characterize diseases
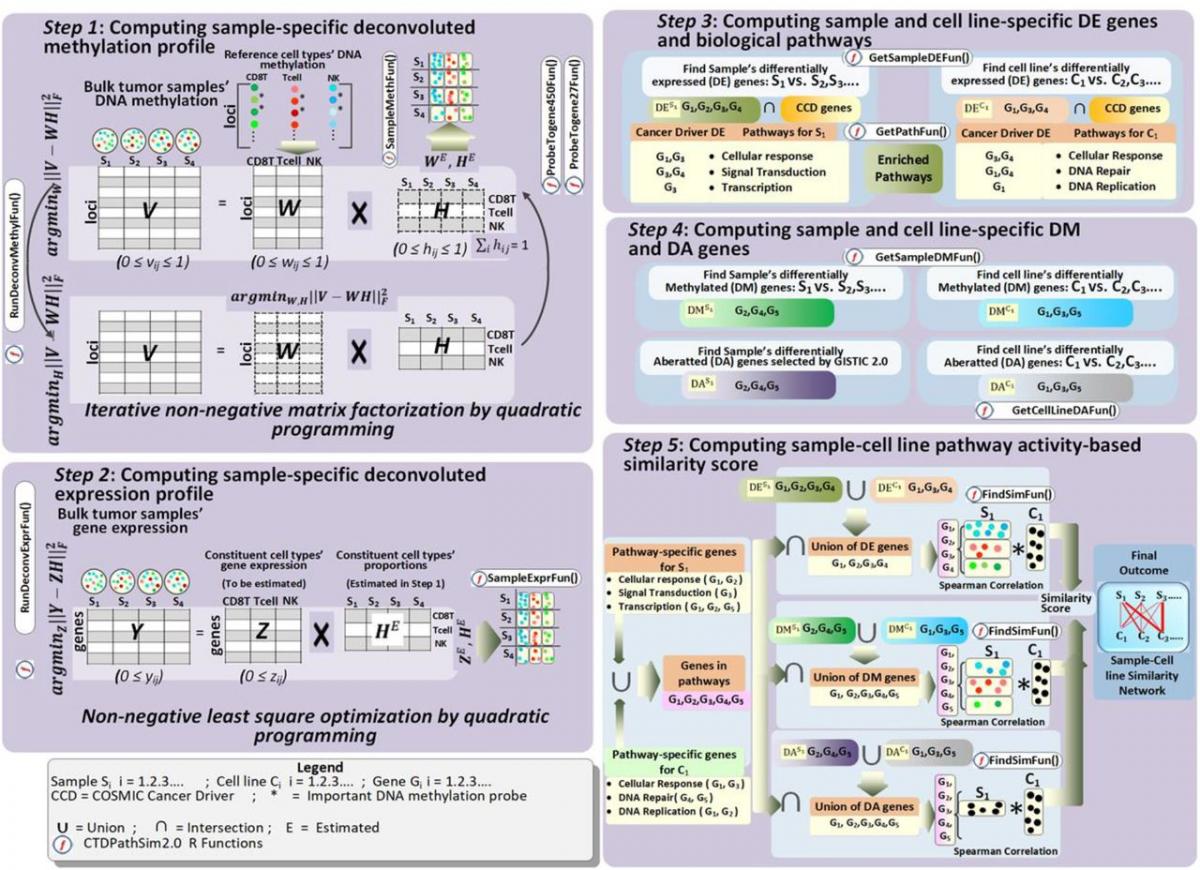
We apply statistical and data mining methods to analyze high dimensional biological datasets to better characterize the diseases. To this end, we have developed CNAReporter software, which is a tool to analyze and report genome-wide genomic alterations, GliomaPredict, a tool that classifies glioma patients into molecular subtypes based on gene expression, and an R/Bioconductor package rgsepd to analyze RNA-seq datasets to perform differential expression and enrichment analysis, and clustering. We analyzed copy number alteration, DNA methylation, mutation and gene expression datasets in glioblastoma patients to identify age-specific genomic, genetic and epigenetic signatures in glioblastoma. In another project, we studied the effect of DNA methylation on mRNA expression of patient-derived glioma stem cells. Most recently, we have conducted an extensive study to compute cancer cell line and primary tumor tissue similarity across pan-cancer.
Selected Publications
Ma Y, Ayadhury S, Singh S, Vashishath Y, Ozdemir C, McKee T, Nguyen N, Basi A, Mak D, Gomez J, Huse J, Noor S, Winkowski D, Baird R, Weinberg J, Lang F, Burks J, Bozdag S, Seeley E, Ene C. Integrated single cell spatial multi-omics landscape of WHO grades 2-4 diffuse gliomas identifies locoregional metabolomic regulators of glioma growth. bioRxiv; 2025. p. 2025.04.30.651361. https://www.biorxiv.org/content/10.1101/2025.04.30.651361v1
S. S. Ferreira, S. Pandey, J. Hemminger, S. Bozdag, and M. S. Antunes, “Early changes in microRNA expression in Arabidopsis plants infected with the fungal pathogen Fusarium graminearum,” PLoS One, vol. 20, no. 2, p. e0318532, Feb. 2025, https://doi.org/10.1371/journal.pone.0318532.
Bose B, Bozdag S. Finding the best cell lines across pan-cancer to use in pre-clinical research as a proxy for patient tumor samples considering immune cells, multi-omics, and cancer pathways. bioRxiv; 2022 https://doi.org/10.1101/2022.12.18.520831
Stamm, K., Tomita-Mitchell, A., & Bozdag, S. (2019). GSEPD: a Bioconductor package for RNA-seq gene set enrichment and projection display. BMC Bioinformatics, 20(1), 115. https://doi.org/10.1186/s12859-019-2697-5
Ready, D., Yagiz, K., Amin, P., Yildiz, Y., Funari, V., Bozdag, S., & Cinar, B. (2017). Mapping the STK4/Hippo signaling network in prostate cancer cell. PLoS ONE, 12(9). https://doi.org/10.1371/journal.pone.0184590
Baur, B., & Bozdag, S. (2017). ProcessDriver: A computational pipeline to identify copy number drivers and associated disrupted biological processes in cancer. Genomics, 109(3-4), 233-240. https://doi.org/10.1016/j.ygeno.2017.04.004
Bozdag, S., Li, A., Riddick, G., Kotliarov, Y., Baysan, M., Iwamoto, F. M., … Fine, H. A. (2013). Age-Specific Signatures of Glioblastoma at the Genomic, Genetic, and Epigenetic Levels. PLoS ONE, 8(4). https://doi.org/10.1371/journal.pone.0062982
Wuchty, S., Arjona, D., Bozdag, S., & Bauer, P. O. (2012). Involvement of microRNA families in cancer. Nucleic Acids Res, 40(17), 8219-8226. https://doi.org/10.1093/nar/gks627
4. Reverse engineering of gene regulatory networks
Reverse engineering of gene regulatory networks is an important and challenging task in computational biology. We have contributed to this research area by developing several tools. Most recently we developed Cancerin and Crinet to infer cancer-specific competing endogenous RNA interactions by integrating various multi-omics datasets and miRDriver to infer miRNA-gene regulatory interactions in pan-cancer. We also implemented a canonical correlation analysis-based algorithm that integrates DNA methylation and copy number alteration with gene expression to infer gene regulatory interactions. We also develop FastMEDUSA , a parallelized version of MEDUSA to infer gene regulatory networks.
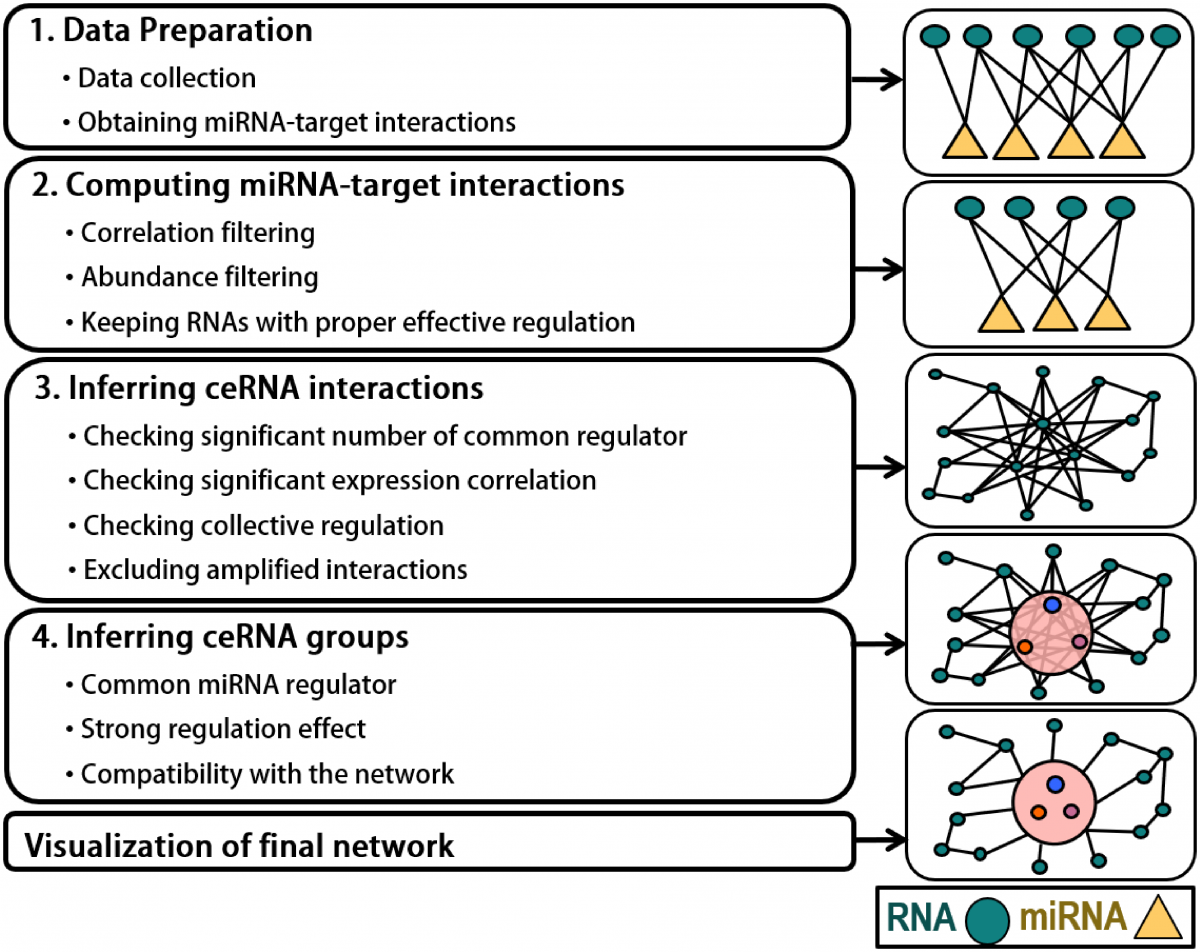
Selected Publications
Z. N. Kesimoglu, J. I. M. Rifat, and S. Bozdag, “Computational inference of co-regulatory modules from transcription factors, MicroRNAs, and their targets using CanMod2,” Sci Rep, vol. 15, no. 1, p. 12521, Apr. 2025, https://doi.org/10.1038/s41598-025-97476-4.
Bose B, Moravec M, Bozdag S. Computing microRNA-gene interaction networks in pan-cancer using miRDriver. Sci Rep. Nature Publishing Group; 2022 Mar 8;12(1):3717. https://doi.org/10.1038/s41598-022-07628-z
Kesimoglu ZN, Bozdag S. Crinet: A computational tool to infer genome-wide competing endogenous RNA (ceRNA) interactions. PLoS One. 2021;16(5):e0251399. PMCID: PMC8118266 https://doi.org/10.1371/journal.pone.0251399
Do D, Bozdag S. CanMod: A computational model to identify co-regulatory modules in cancer. Proceedings of the 11th ACM International Conference on Bioinformatics, Computational Biology and Health Informatics . New York, NY, USA: Association for Computing Machinery; 2020. p. 1-10. https://doi.org/10.1145/3388440.3415586
Do, D., & Bozdag, S. (2018). Cancerin: A computational pipeline to infer cancer-associated ceRNA interaction networks. PLoS Computational Biology, 14(7), e1006318. https://doi.org/10.1371/journal.pcbi.1006318
Baur, B., & Bozdag, S. (2015). A canonical correlation analysis-based dynamic bayesian network prior to infer gene regulatory networks from multiple types of biological data. Journal of Computational Biology: A Journal of Computational Molecular Cell Biology, 22(4), 289-299. https://doi.org/10.1089/cmb.2014.0296
Bozdag, S., Li, A., Wuchty, S., & Fine, H. A. (2010). FastMEDUSA: a parallelized tool to infer gene regulatory networks. Bioinformatics, 26(14), 1792-1793. https://doi.org/10.1093/bioinformatics/btq275
